A brief course of Molecular evolution for biologistsⅠ
Yuliang Zhang / 2017-10-13
The most exciting part of biology is evolution theory, and the main research content of modern evolution research is molecular evolution, include sequences alignment, phylogenetics inference, and so on. Here, I will write a series of blogs to explain the basic consepts of molecular evolution, aslo, as notes of my own learning process. I am a biologist, so these blogs are biologists friendly, I will try my best to not involve too much mathematical formulas. Let’s take the frist step:
Evolution Distance
Evolution distance(D) is denfined as difference between two homologous sequences. we consider there are two sequences: Seq1, Seq2 from two spices originate from a ancestral sequence Seq-Ance before t time units ago, as the below figure shows:
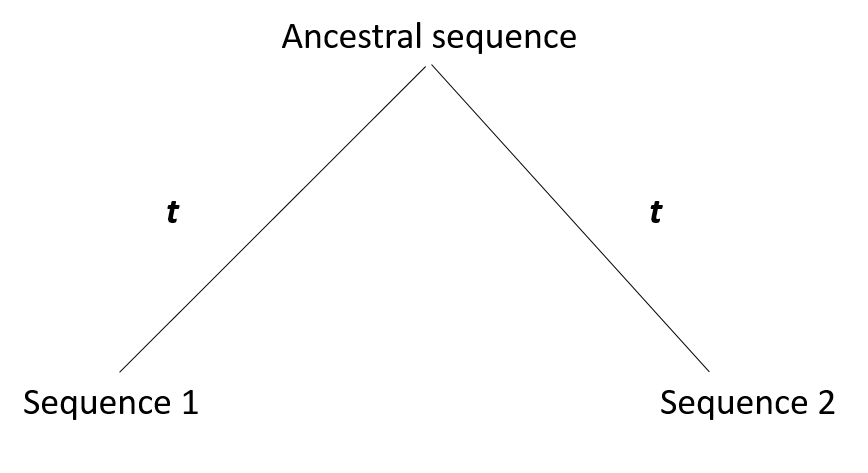 For example, the ancestral sequence is:
For example, the ancestral sequence is:
Seq-Ance:
AATCGGCTACTAGCT1.
after t time units, Sequence1 from spice1 has 4 base substitutions and Sequence2 from spice2 has 6 base substitutions:
Sequence1:
ATTCCGCTGCTAACT
sequence2:
TAGCAGCCACTGCCT
Then evolution distance(D) can be calculated as fellows: $$D=\frac{4+6}{15}=\frac{2} {3}$$
But we could never actually know how many substitutions each of the sequences has undergone2, so next we discuss estimating D from two sequences.
Estimate Evolution distance
There are various types of DNA regions such as protein-coding regions, no-coding regions, introns, exons and so on. Some regions maybe under the pressure of selection, here we consider two simple regions, for example, introns and protein-coding regions.
The uniform rate model for Introns
In no-coding regions(like introns), it is reasonable to assume that all sites(base) in the sequence follow the same substitution model and evolve at the same rate. Frist we discuss the Jukes-Cantor Model: we consider a substitution rate martix:
| base | A | T | G | C |
|---|---|---|---|---|
| A | 1-3μ | μ | μ | μ |
| T | μ | 1-3μ | μ | μ |
| G | μ | μ | 1-3μ | μ |
| C | μ | μ | μ | 1-3μ |
this model assume that on a given site the substitution rate(probability) is μ(eg.5*10-9 (per site)/(per year)), then the probability p(t) that two homologous sequences are different at that site at time t is given by: $$p_{diff}(t)= \frac{3}{4} (1-e^{-8\mu t}) $$
and the evolution distance is given by: $$D=-\frac{3}{4}ln(1-\frac{4}{3}p_{diff}(t))$$
There is the proof, if you don’t want konw all the details or unfamiliar with statistics, you can just skip this part.
Proof
If the ancestor sequence has base x at a given site, then the two descent sequences would be the same at that site if the base did not change in both of them or if both bases changed to the same base.
Frist we consider one sequence, if a base at a given site did not change at time t, there are two possibilities:
- the base did not change
- and base changed to another base from x at time t-1 and then change to x at time t
Let P(x|x,t) be the probability that after time t time unit the site still has base x. let p(t)=P(x|x,t), then:
$$ p(t+1)=\sum_y P(site=x\ at\ time\ t+1|site = y\ at\ time\ t)P(site=y\ at\ time\ t)$$ $$=\mu \frac{1-p(t)}{3} + \mu \frac{1-p(t)}{3} + \mu \frac{1-p(t)}{3} +(1-3\mu)p(t)$$ $$=p(t)(1-4\mu) +\mu $$
if we let $c=1-4μ$, we then have that
$$p(t+1)=cp(t)+\mu$$
we solve this recurrence as follows: $$ p(0)=1 $$ $$p(1)=c(1)+\mu=c+\mu $$ $$ p(2)=c(c+\mu)+\mu=c^2+(c+1)\mu $$ $$ p(t)=c^t+(c^{t-1}+c^{t-2}+…+1)\mu =c^t+\frac{c^t-1}{c-1}\mu=\frac{1}{4}+\frac{3}{4}(1-4\mu)^t $$
then the \(P(y|x,t) (x\ not\ equal to y)=(1-P(x|x,t))/3 \),if $\mu$ is small $1-4\mu \approx e^{-4\mu t}$
So the probability r(t) that two sequences are the same at a given site at time t is given by3: $$ r(t)=\sum_y{[P(y|x,t)]^2}=[P(x|x,t)]^2+[P(y|x,t)]^2=\frac{1}{4}+\frac{3}{4}e^{-8\mu t}$$
So, the probability that two homologous sequences are different at a given site at time t is given by: $$ p_{diff}(t)=1-r(t)=\frac{3}{4}(1-e^{-8\mu t})$$
if the length of the sequence is n, then the evolution distance:
$$ D=2\frac{3\mu t \times n}{n}=6\mu t$$
we can obtain the relationship between $p_{diff}(t)$ and $\mu$:
$$\mu = \frac{-ln(1-\frac{4}{3}p_{diff}(t))}{8t}$$
so the evolution distance is given by:
$$D=-\frac{3}{4}ln(1-\frac{4}{3}p_{diff}(t))$$
This completes the proof.
The Nonuniform rate model for protein-coding regions
I won’t discuss the details of the Nonuniform rate model. In protein-coding regions, it is not reasonable to assume that all site have the same evole rate, but we can aslo given a calculate formula of evolution distance.
An example for calaulating the D
as we mentioned before, there are two homologous sequences: Sequence1 from spice1 has 4 base substitutions and Sequence2 from spice2 has 6 base substitutions:
Sequence1:
ATTCCGCTGCTAACT
sequence2:
TAGCAGCCACTGCCT
after the alignment, we found there are 8 different sites, so the pdiff=8⁄15, so the evolution distance is calculated as 0.9313.
The Jukes-Cantor Model is the simplest model of evolution distance estimation, other models can be found at Masatoshi Nei’s book 《Molecular Evolution and Phylogenetics》, now we can calculate the evolution distance between any two homologous sequences, we will use the evolution distance to build a phylogenetic tree in next bolg.